Transcription factor regulation as a mechanism of confounding effects between distinct human traits
Abstract
Corresponding author: Milos Pjanic
How to cite: Pjanic M, Miller CL and Quertermous T. Transcription factor regulation as a mechanism of confounding effects between distinct human traits [version 1; referees: 1 not approved]. F1000Research 2015, 4:1349 (doi: 10.12688/f1000research.7336.1)Copyright: © 2015 Pjanic M et al. This is an open access article distributed under the terms of the Creative Commons Attribution Licence, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The author(s) is/are employees of the US Government and therefore domestic copyright protection in USA does not apply to this work. The work may be protected under the copyright laws of other jurisdictions when used in those jurisdictions.
Competing interests:
The authors have no conflicts of interest or competing interests to disclose.
First published: 25 Nov 2015, 4:1349 (doi: 10.12688/f1000research.7336.1)Latest published: 25 Nov 2015, 4:1349 (doi: 10.12688/f1000research.7336.1)Introduction
A recent publication1 has demonstrated the potential causative mechanism of a genome-wide association study (GWAS) locus for the development of blond hair. Through a series of elegant in vivo experiments in mice, the study's findings strengthen association of single nucleotide polymorphism (SNP) rs12821256, initially discovered as one of the top GWAS hits in European populations2, with light hair color development. This work implicates a mechanism of long-range regulation of a gene on chromosome 12, termed KITLG that encodes the ligand for a receptor-type protein-tyrosine kinase, and is located 350kb away from the variant. Further, using data generated by the ENCODE consortium, the study reveals a molecular mechanism by which SNP rs12821256 confers the blond hair phenotype via directly altering a canonical binding site for transcription factor TCF7L23. This may shed light on possible cis- and trans-acting mechanisms responsible for the association of rs12821256 with the quantitative trait of light hair color.
On the other hand, the TCF7L2 locus on chromosome 10 is well-known for its strong association with type 2 diabetes(T2D) and glycemic traits from several GWAS studies4,5. It confers the strongest effect on T2D to date, with a per-allele odds ratio of 1.396. Lead risk-associated SNPs from the TCF7L2 locus include two intronic SNPs (rs7903146 and rs4506565). The majority of SNPs from the TCF7L2 locus are non-coding and may alter the levels of expression or affect alternative splicing of TCF7L2, while SNPs located in TCF7L2 exons give rise to alternate protein isoforms. In addition, numerous SNPs from this locus that are in linkage disequilibrium (LD) with GWAS lead SNPs could be candidates for the causal variant(s). Given these reports, it seems likely that specific TCF7L2 expression levels or the composition of its 13 or more transcripts (UCSC annotation) and isoforms in pancreatic beta cells confer risk for T2D, while in melanocytes the composition of TCF7L2 variants and levels may influence trans TCF7L2 protein binding to SNP rs12821256 to alter expression of the downstream KITLG gene, an important regulator of melanogenesis.
TCF7L2 is expressed in a variety of human tissues, where it plays a critical role in the Wnt signaling pathway. In skin tissues TCF7L2 reaches moderate expression levels with RPKM (Reads Per Kilobase of transcript per Million mapped reads) values between 10 and 20, which are higher than that observed in pancreas (<10)7.
Main body
In addition to binding to rs12821256, we report here that TCF7L2 binds to the promoter region of the KITLG gene (as shown in the ENCODE ChIP-Seq data sets), as well as throughout the first intron and immediate upstream region, and overlaps the active enhancer histone modification mark H3K27ac (Figure 1A), which further implicates its role in the regulation of KITLG expression. When we queried the Genotype-Tissue Expression (GTEx) database or eQTL resources from the Gilad/Pritchard group there were no SNPs from the TCF7L2 locus detected as expression quantitative trait loci (eQTL SNPs) for KITLG (search terms in Supplementary Table S1), nor when we investigated HapMap data through the GENEVAR (GENe Expression VARiation) platform. However, a significant eQTL association between TCF7L2 SNPs (rs7903146 and rs12255372) and KITLG was observed in skin tissues in data from the MuTHER (Multiple Tissue Human Expression Resource) healthy female twin studies8 (Figure 1B, p=0.0089 and 0.0349, respectively), implicating a strong trans-eQTL interaction in skin tissues compared to Lymphoblastoid cell lines (LCL) or adipose tissues where either the absence of, or weak eQTL association was found.
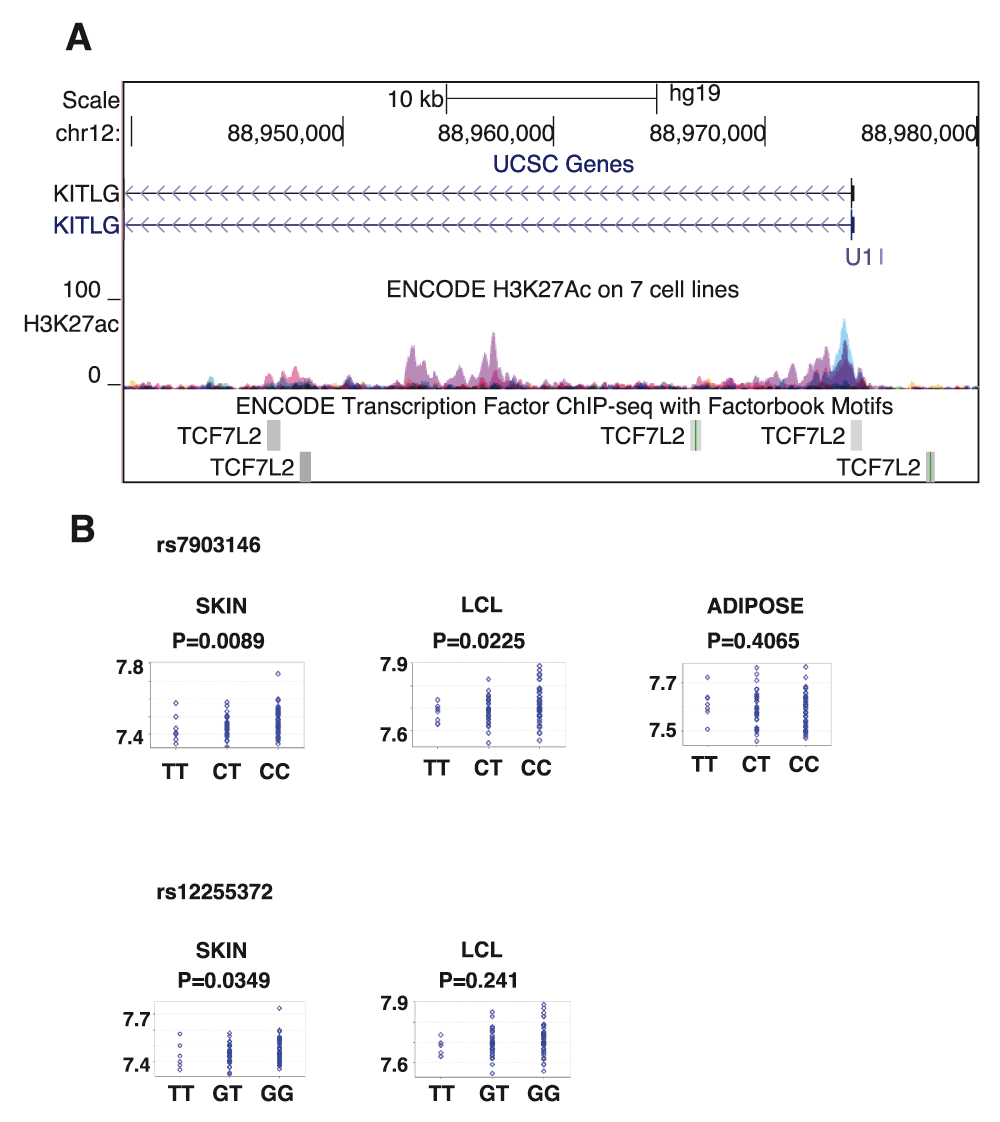
Figure 1. TCF7L2 locus variation and protein occupancy implicated in regulation of KITLG gene.
A. TCF7L2 protein binding at the KITLG promoter, upstream of the KITLG promoter and multiple binding events in the first intron of KITLG gene. TCF7L2 binding sites overlap regulatory histone mark H3K27ac, implying their functionality in gene regulation. Data were taken from the ENCODE consortium. B. eQTL analysis of two T2D SNPs from TCF7L2 locus (rs7903146 and rs12255372) and KITLG gene in multiple tissues: skin, lymphoblastoid cell line (LCL) and adipose. Data from MuTHER healthy female twin studies.
As demonstrated by the International Diabetes Federation data for 20149, T2D is less prevalent in northern European populations (compared to, e.g. southern Europeans). Data of the frequency of T2D patients in Europe shows that southern European countries, i.e. Spain (7.9%), Portugal (9.6%), Balkan countries (9.8%) and Turkey (14.8%) have the highest percentage of T2D patients in Europe (average 10.2%). On the other hand, northern European countries like Britain (3.9%), Sweden (4.5%), Norway (5.2), Baltic countries (3.8%, 5.0% and 5.7%), and Iceland (3.2%) have lower percentage of diabetics compared to the rest of Europe (average 3.9%). This difference in disease prevalence could be attributed to differences in dietary or other environmental factors, but also could reflect differences in allele frequency of disease-associated alleles. In fact, the frequencies of SNP rs12821256 and light hair color are more common in northern European populations, e.g., blond and light brown hair reaching 75% in Icelandic populations and rs12821256 MAF reaching its frequency maximum of 0.19 in Iceland (Supplementary Figure S1)1,2. Similarly, using data from ALFRED (ALlele FRequency Database), we found an inverse correlation of population’s geographic latitude and frequency of TCF7L2 SNP rs7903146 (Figure 2A), and another TCF7L2 SNP rs12255372 also showed a similar trend (Figure 2B). Thus, it is intriguing to speculate whether TCF7L2 protein isoforms may give rise to light hair color via binding to rs12821256 and regulating the KITLG gene in one cell type (melanocytes), while in pancreatic beta cells they may act as risk factors for the development of diabetes (Figure 3), through TCF7L2 gene regulation and potential cross-composition of TCF7L2isoforms.
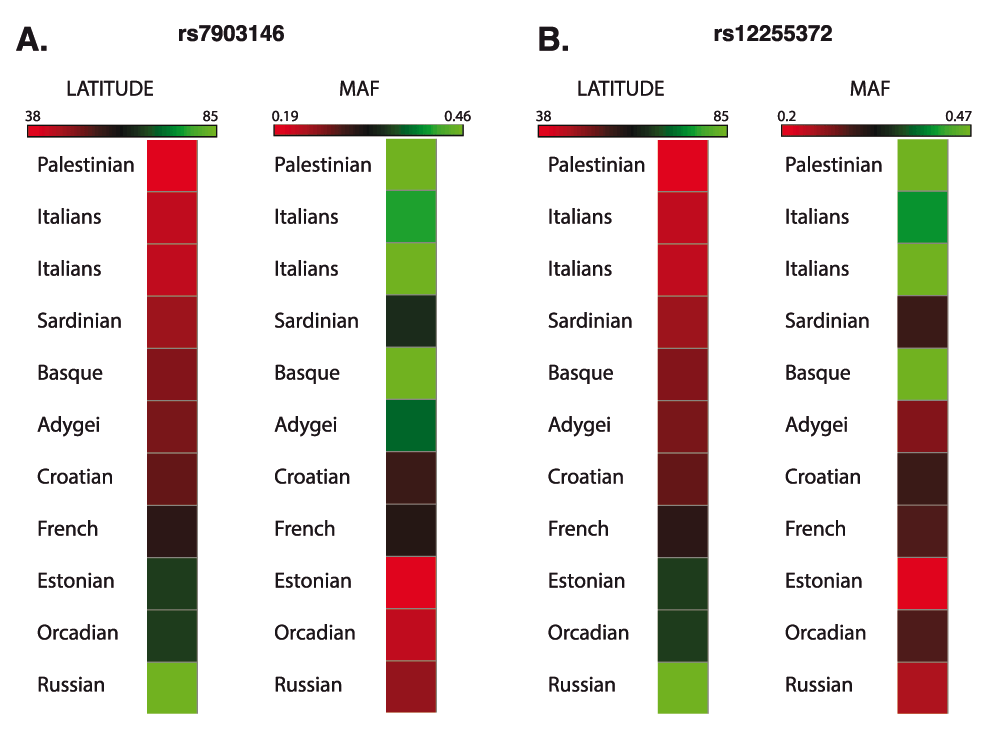
Figure 2. Inverse correlation of geographic latitude and T2D SNP minor allele frequency.
Maximal geographical latitude of the population and T2D SNP minor allele frequency (MAF) were taken from Alfred (Allele Frequency Database) and plotted as a heatmap. A. SNP rs7903146 B. SNP rs12255372.
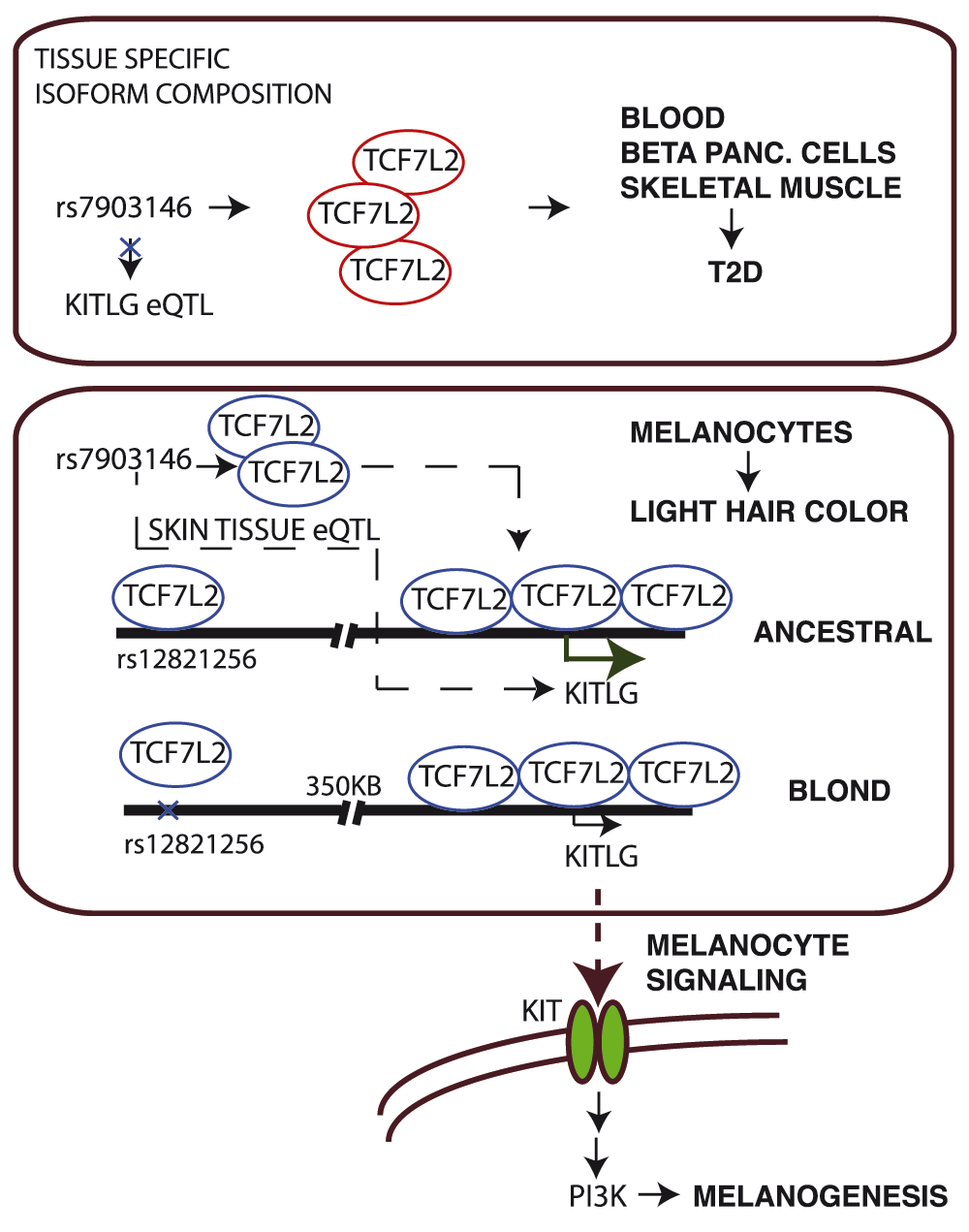
Figure 3. Schematic representation of transcription factor regulation as basis for confounding effects between diseases and traits.
T2D SNP rs7903146 from TCF7L2 locus is shown as eQTL SNP for KITLG gene in skin tissues. eQTL association is lost in other tissues, indicating regulation of KITLG gene by TCF7L2 isoforms explicitly in skin tissues.
Conclusion
In summary, the putative trans-eQTL interaction in skin tissues we report here implicates natural genetic variation in the T2D locus, TCF7L2, to regulate expression of KITLG, a gene linked to light hair color development. We postulate that this could be the underlying genetic mechanism accounting for the association between hair color and T2D risk in European populations. Our observations here strengthen the hypothesis of a genetically determined correlation between diseases and traits in human population, as also demonstrated in a recent publication with the inversely correlated height and coronary artery disease (CAD) phenotypes, where height-associated variants were associated with an increase of 13.5% in the risk of CAD10. Furthermore, these observations illustrate how investigating the genetic architecture underlying complex traits and diseases may inform appropriate risk stratification in diverse human populations.


Comments
Post a Comment